Offering multiple perspectives from many fields of human inquiry that may move all of us toward a more integrated understanding of who we are as conscious beings.
From the NIH, this is a nearly 7 hour video of a recent conference on the cellular of molecular mechanisms of physical activity-induced health benefits (i.e. prevents disease or improves overall health).
Description: The NIH Common Fund is currently exploring research needs and opportunities related to the molecular mechanisms whereby physical activity prevents disease and improves health outcomes. This activity is undertaken with the leadership of the NIH Institute Directors Richard Hodes, M.D., National Institute on Aging (NIA), Stephen I. Katz, M.D., Ph.D., National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), and Griffin Rodgers, M.D., National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and with broad support throughout the NIH. The Trans NIH Committee Physical Activity Common Fund (PACF) Working Group plans to cover the broad aspects of physical activity related benefits under 5 sub-working groups within these ICs (NIAMS, NIDDK, NIA and OSC) Download: To download this event, select one of the available bitrates: [64k][150k][240k][440k][740k][1040k]How to download a Videocast Caption Text: Download Caption File
From Nautilus, this is a very cool article by Ed Yong (blogger at the National Geographic's Phenomena: Not Exactly Rocket Science blog) on the emergence of eukaryotic cells, an enormous leap in complexity and arguably one of the "most important event in the history of life on Earth."
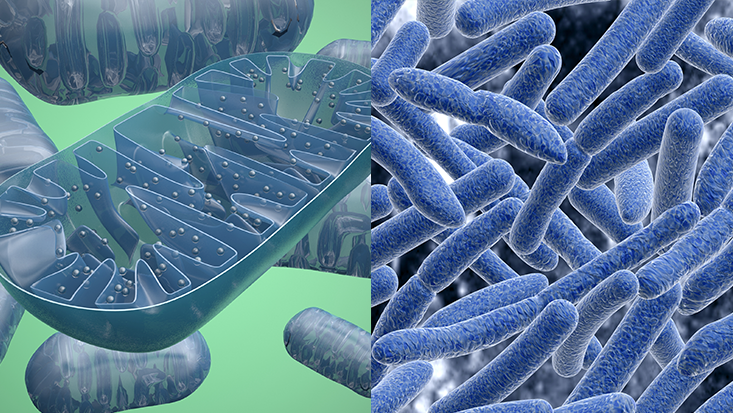
The emergence of eukaryotic life also brought along with it the emergence of mitochondria, the cell's "power plant" in that it generates most of the adenosine triphosphate (ATP) used for energy.
Interestingly, unlike other evolutionary innovations, the eukaryotic cell and mitochondria only appear once in the evolutionary timeline. Fortunately for us, they stuck around.
All sophisticated life on the planet Earth may owe its existence to one freakish event.
By Ed Yong | September 4, 2014
ILLUSTRATION BY GRACIA LAM
AT FIRST GLANCE, a tree could not be more different from the caterpillars that eat its leaves, the mushrooms sprouting from its bark, the grass growing by its trunk, or the humans canoodling under its shade. Appearances, however, can be deceiving. Zoom in closely, and you will see that these organisms are all surprisingly similar at a microscopic level. Specifically, they all consist of cells that share the same basic architecture. These cells contain a central nucleus—a command center that is stuffed with DNA and walled off by a membrane. Surrounding it are many smaller compartments that act like tiny organs, carrying out specialized tasks like storing molecules or making proteins. Among these are the mitochondria—bean-shaped power plants that provide the cells with energy. This combination of features is shared by almost every cell in every animal, plant, fungus, and alga, a group of organisms known as “eukaryotes.” Bacteria showcase a second, simpler way of building a cell—one that preceded the complex eukaryotes by at least a billion years. These “prokaryotes” always consist of a single cell, which is smaller than a typical eukaryotic one and bereft of internal compartments like mitochondria and a nucleus. Even though limited to a relatively simple cell, bacteria are impressive survival machines. They colonize every possible habitat, from miles-high clouds to the deep ocean. They have a dazzling array of biological tricks that allow them to cause diseases, eat crude oil, conduct electric currents, draw power from the Sun, and communicate with each other. Still, without the eukaryotic architecture, bacteria are forever constrained in size and complexity. Sure, they have their amazing skill sets, but it’s the eukaryotes that cover the Earth in forest and grassland, that navigate the planet looking for food and mates, that build rockets to Mars. The transition from the classic prokaryotic model to the deluxe eukaryotic one is arguably the most important event in the history of life on Earth. And in more than 3 billion years of existence, it happened exactly once. Life is full of complex structures that evolve time and again. Individual cells have united to form many-celled creatures like animals and plants on dozens of separate occasions. The same is true for eyes, which have independently evolved time and again. But the eukaryotic cell is a one-off innovation. Bacteria have repeatedly nudged along the path towards complexity. Some are very big (for microbes); others move in colonies that behave like single, many-celled creatures. But none of them have acquired the full suite of crucial features that define eukaryotes: large size, the nucleus, internal compartments, mitochondria, and more. As Nick Lane from University College London writes, “Bacteria have made a start up every avenue of eukaryotic complexity, but then stopped short.” Why? It is not for lack of opportunity. The world is swarming with countless prokaryotes that evolve at breathtaking rates. Even so, they were not quick about inventing eukaryotic cells. Fossils tell us that the oldest bacteria arose between 3 and 3.5 billion years ago, but there are no eukaryotes from before 2.1 billion years ago. Why did the prokaryotes remain as simple cells for so damn long? There are many possible explanations, but one of these has recently gained a lot of ground. It tells of a prokaryote that somehow found its way inside another, and formed a lasting partnership with its host. This inner cell—a bacterium—abandoned its free-living existence and eventually transformed into the mitochondria. These internal power plants provided the host cell with a bonanza of energy, allowing it to evolve in new directions that other prokaryotes could never reach. If this story is true, and there are still those who doubt it, then all eukaryotes—every flower and fungus, spider and sparrow, man and woman—descended from a sudden and breathtakingly improbable merger between two microbes. They were our great-great-great-great-...-great-grandparents, and by becoming one, they laid the groundwork for the life forms that seem to make our planet so special. The world as we see it (and the fact that we see it at all; eyes are a eukaryotic invention) was irrevocably changed by that fateful union—a union so unlikely that it very well might not have happened at all, leaving our world forever dominated by microbes, never to welcome sophisticated and amazing life like trees, mushrooms, caterpillars, and us.
IN 1905, the Russian biologist Konstantin Mereschkowski first suggested that some parts of eukaryotic cells were once endosymbionts—free-living microbes that took up permanent residence within other cells. He thought the nucleus originated in this way, as did the chloroplasts that allow plant cells to harness sunlight. He missed the mitochondria, but the American anatomist Ivan Wallin pegged them for endosymbionts in 1923. These ideas were ignored for decades until an American biologist—the late Lynn Margulis—revived them in 1967. In a radical paper, she made the case that mitochondria and chloroplasts were once free-living bacteria that had been sequentially ingested by another ancient microbe. That is why they still have their own tiny genomes and why they still superficially look like bacteria. Margulis argued that endosymbiosis was not a crazy, oddball concept—it was one of the most important leitmotivs in the eukaryotic opera. The paper was a tour de force of cell biology, biochemistry, geology, genetics, and paleontology. Its conclusion was also grossly unorthodox. At the time, most people believed that mitochondria had simply come from other parts of the cell.
“[Endosymbiosis] was taboo,” says Bill Martin from Heinrich Heine University Düsseldorf, in Germany. “You had to sneak into a closet to whisper to yourself about it before coming out again.” Margulis’ views drew fierce criticism, but she defended with equal vigor. Soon she had the weight of evidence behind her. Genetic studies, for example, showed that mitochondrial DNA is similar to that of free-living bacteria. Now, very few scientists doubt that mergers infused the cells of every animal and plant with the descendants of ancient bacteria. But the timing of that merger, the nature of its participants, and its relevance to the rise of eukaryotes are all still hotly debated. In recent decades,origin stories for the eukaryotes have sprouted up faster than old ones could be tested, but most fall into two broad camps. The first—let’s call it the “gradual-origin” group—claimed that prokaryotes evolved into eukaryotes by incrementally growing in size and picking up traits like a nucleus and the ability to swallow other cells. Along the way, these proto-eukaryotes gained mitochondria, because they would regularly engulf bacteria. This story is slow, steady, and classically Darwinian in nature. The acquisition of mitochondria was just another step in a long, gradual transition. This is what the late Margulis believed right till the end. The alternative—let’s call it the “sudden-origin” camp—is very different. It dispenses with slow, Darwinian progress and says that eukaryotes were born through the abrupt and dramatic union of two prokaryotes. One was a bacterium. The other was part of the other great lineage of prokaryotes: the archaea. (More about them later.) These two microbes look superficially alike, but they are as different in their biochemistry as PCs and Macs are in their operating systems. By merging, they created, in effect, the starting point for the first eukaryotes. Bill Martin and Miklós Müller put forward one of the earliest versions of this idea in 1998. They called it the hydrogen hypothesis. It involved an ancient archaeon that, like many modern members, drew energy by bonding hydrogen and carbon dioxide to make methane. It partnered with a bacterium that produced hydrogen and carbon dioxide, which the archaeon could then use. Over time, they became inseparable, and the bacterium became a mitochondrion. There are many variants of this hypothesis, which differ in the reasons for the merger and the exact identities of the archaeon and the bacterium that were involved. But they are all united by one critical feature setting them apart from the gradual-origin ideas: They all say that the host cell was still a bona fide prokaryote. It was an archaeon, through and through. It had not started to grow in size. It did not have a nucleus. It was not on the path to becoming a eukaryote; it set off down that path because it merged with a bacterium. As Martin puts it, “The inventions came later.” This distinction could not be more important. According to the sudden-origin ideas, mitochondria were not just one of many innovations for the early eukaryotes. “The acquisition of mitochondria was the origin of eukaryotes,” says Lane. “They were one and the same event.” If that is right, the rise of the eukaryotes was a fundamentally different sort of evolutionary transition than the gradual changes that led to the eye, or photosynthesis, or the move from sea to land. It was a fluke event of incredible improbability—one that, as far as we know, only happened after a billion years of life on Earth and has not been repeated in the 2 billion years since. “It’s a fun and thrilling possibility,” says Lane. “It may not be true, but it’s beautiful.” IN 1977, microbiologist Carl Woese had the bright idea of comparing different organisms by sequencing their genes. This is an everyday part of modern biology, but at the time, scientists relied on physical traits to deduce the evolutionary relationships between different species. Comparing genes was bold and new, and it would play a critical role in showing how complicated life like us—the eukaryotes—came to be. Woese focused on 16S rRNA, a gene that is involved in the essential task of making proteins and is found in all living things. Woese reasoned that as organisms diverge into new species, their versions of rRNA should become increasingly dissimilar. By comparing the gene across a range of prokaryotes and eukaryotes, the branches of the tree of life should reveal themselves. They did, but no one expected the results. Woese’s tree had three main branches. Bacteria and eukaryotes sat on two of them. But the third consisted of an obscure bunch of prokaryotes that had been found in hot, inhospitable environments. Woese called them archaea, from the Greek word for ancient. Everyone had taken them for obscure types of bacteria, but Woese’s tree announced them as a third domain of life. It was as if everyone was staring at a world map, and Woese had politely shown that a full third of it had been folded underneath. In Woese’s classic three-domain tree, the eukaryotes and archaea are sister groups. They both evolved from a shared ancestor that split off from the bacteria very early in the history of life on Earth. But this tidy picture started to unravel in the 1990s, as the era of modern genetics kicked into high gear and scientists started sequencing more eukaryotic genes. Some were indeed closely related to archaeal genes, but others turned out to be more closely related to bacterial ones. The eukaryotes turned out to be a confusing hodgepodge, and their evolutionary affinities kept on shifting with every new sequenced gene. In 2004, James Lake changed the rules of engagement. Rather than looking at any single gene, he and his colleague Maria Rivera compared the entire genomes of two eukaryotes, three bacteria, and three archaea. Their analysis supported the merger-first ideas: They concluded that the common ancestor of all life diverged into bacteria and archaea, which evolved independently until two of their members suddenly merged. This created the first eukaryotes and closed what now appeared to be a “ring of life.” Before that fateful encounter, life had just two major domains. Afterward, it had three. Rivera and Lake were later criticized for only looking at seven species, but no one could possibly accuse Irish evolutionary biologist James McInerney of the same fault. In 2007, he crafted a super-tree using more than 5,700 genes from across the genomes of 168 prokaryotes and 17 eukaryotes. His conclusion was the same: Eukaryotes are merger organisms, formed through an ancient symbiosis between a bacterium and an archaeon. The genes from these partners have not integrated seamlessly. They behave like immigrants in New York’s Asian and Latino communities, who share the same city but dominate different areas. For example, they mostly interact with their own kind: archaeal genes with other archaeal genes, and bacterial genes with bacterial genes. “You’ve got two groups in the playground and they’re playing with each other differently, because they’ve spent different amounts of time with each other,” says McInerney. They also do different jobs. The archaeal genes are more likely to be involved in copying and making use of DNA. The bacterial genes are more involved in breaking down food, making nutrients, and the other day-to-day aspects of being a microbe. And although the archaeal genes are outnumbered by their bacterial neighbors by 4 to 1, they seem to be more important. They are nearly twice as active. They produce proteins that play more central roles in their respective cells. They are more likely to kill their host if they are mistakenly deleted. Over the last four years, McInerney has found this same pattern again and again, in yeast, in humans, in dozens of other eukaryotes. This all makes sense if you believe the sudden-origin idea. When those ancient partners merged, the immigrant bacterial genes had to be integrated around a native archaeal network, which had already been evolving together for countless generations. They did integrate, and while many of the archaeal genes were displaced, an elite set could not be ousted. Despite 2 billion years of evolution, this core network remains, and retains a pivotal role out of all proportion to their small number. THE SUDDEN-ORIGIN hypothesis makes one critical prediction: All eukaryotes must have mitochondria. Any exceptions would be fatal, and in the 1980s, it started to look like there were exceptions aplenty. If you drink the wrong glass of water in the wrong part of the world, your intestines might become home to a gut parasite called Giardia. In the weeks that follow, you can look forward to intense stomach cramps and violent diarrhea. Agony aside, Giardia has a bizarre and interesting anatomy. Itconsists of a single cell that looks like a malevolent teardrop with four tail-like filaments. Inside, it has not one nucleus but two. It is clearly a eukaryote. But it has no mitochondria.
Mitochondria (left) are domesticated versions of bacteria (right) that now provide the cells of every animal, plant and fungus with energy. ShutterstockThere are at least a thousand other single-celled eukaryotes, mostly parasites, which also lack mitochondria. They were once called archezoans, and their missing power plants made them focal points for the debate around eukaryotic origins. They seemed to be living remnants of a time when prokaryotes had already turned into primitive eukaryotes, but before they picked up their mitochondria. Their very existence testified that mitochondria were a late acquisition in the rise of eukaryotes, and threatened to deal a knockout blow to the sudden-origin tales.
That blow was deflected in the 1990s, when scientists slowly realized that Giardia and its ilk have genes that are only ever found in the mitochondria of other eukaryotes. These archezoans must have once had mitochondria, which were later lost or transformed into other cellular compartments. They aren’t primitive eukaryotes from a time before the mitochondrial merger—they are advanced eukaryotes that have degenerated, just as tapeworms and other parasites often lose complex organs they no longer need after they adopt a parasitic way of life. “We’ve yet to find a single primitive, mitochondria-free eukaryote,” says McInerney, “and we’ve done a lot of looking.” With the archezoan club dismantled, the sudden-origin ideas returned to the fore with renewed vigor. “We predicted that all eukaryotes had a mitochondrion,” says Martin. “Everyone was laughing at the time, but it’s now textbook knowledge. I claim victory. Nobody’s giving it to me—except the textbooks.” IF MITOCHONDRIA were so important, why have they only evolved once? And for that matter, why have eukaryotes only evolved once? Nick Lane and Bill Martin answered both questions in 2010, in a bravura paper called, “The energetics of genome complexity,” published in Nature. In a string of simple calculations and elegant logic, they reasoned that prokaryotes have stayed simple because they cannot afford the gas-guzzling lifestyle that all eukaryotes lead. In the paraphrased words of Scotty: They cannae do it, captain, they just don’t have the power. Lane and Martin argued that for a cell to become more complex, it needs a bigger genome. Today, for example, the average eukaryotic genome is around 100–10,0001 times bigger than the average prokaryotic one. But big genomes don’t come for free. A cell needs energy to copy its DNA and to use the information encoded by its genes to make proteins. The latter, in particular, is the most expensive task that a cell performs, soaking up three-quarters of its total energy supply. If a bacterium or archaeon was to expand its genome by 10 times, it would need roughly 10 times more energy to fund the construction of its extra proteins. One solution might be to get bigger. The energy-producing reactions that drive prokaryotes take place across their membranes, so a bigger cell with a larger membrane would have a bigger energy supply. But bigger cells also need to make more proteins, so they would burn more energy than they gained. If a prokaryote scaled up to the same size and genome of a eukaryotic cell, it would end up with 230,000 times less energy to spend on each gene! Even if this woefully inefficient wretch could survive in isolation, it would be easily outcompeted by other prokaryotes. Prokaryotes are stuck in an energetic canyon that keeps them simple and small. They have no way of climbing out. If anything, evolution drives them in the opposite direction, mercilessly pruning their genomes into a ring of densely packed and overlapping genes. Only once did a prokaryote escape from the canyon, through a singular and improbable trick—it acquired mitochondria. Mitochondria have an inner membrane that folds in on itself like heavily ruched fabric. They offer their host cells a huge surface area for energy-producing chemical reactions. But these reactions are volatile, fickle things. They involve a chain of proteins in the mitochondrial membranes that release energy by stripping electrons from food molecules, passing them along to one another, and dumping them onto oxygen. This produces high electric voltages and unstable molecules. If anything goes wrong, the cell can easily die. But mitochondria also have a tiny stock of DNA that encodes about a dozen of the proteins that take part in these electron-transfer chains. They can quickly make more or less of any of the participating proteins, to keep the voltages across their membranes under check. They supply both power and the ability to control that power. And they do that without having to bother the nucleus. They are specialized to harness energy. Mitochondria are truly the powerhouse of the eukaryotic cell. “The command center is too bureaucratic and far away to do anything,” says Lane. “You need to have these small teams, which have limited powers but can use them at their discretion to respond to local situations. If they’re not there, everything dies.” Prokaryotes do not have powerhouses; they are powerhouses. They can fold their membranes inwards to gain extra space for producing energy, and many do. But they do not have the secondary DNA outposts that produce high-energy molecules so the central government (the nucleus) has the time and energy to undertake evolutionary experiments. The only way to do that is to merge with another cell. When one archaeon did so, it instantly leapt out of its energetic canyon, powered by its new bacterial partner. It could afford to expand its genome, to experiment with new types of genes and proteins, to get bigger, and to evolve down new and innovative routes. It could form a nucleus to contain its genetic material, and absorb other microbes to use as new tiny organs, like the chloroplasts that perform photosynthesis in plants. “You need a mitochondrial level of power to finance those evolutionary adventures,” says Martin. “They don’t come for free.” Lane and Martin’s argument is a huge boon for the sudden-origin hypothesis. To become complex, cells need the stable, distributed energy supply that only mitochondria can provide. Without these internal power stations, other prokaryotes, for all their evolutionary ingenuity, have always stayed as single, simple cells. The kind of merger that creates mitochondria seems to be a ludicrously unlikely event. Prokaryotes have only managed it once in more than 3 billion years, despite coming into contact with each other all the time. “There must have been thousands or millions of these cases over evolutionary time, but they’ve got to find a way of getting along, of reconciling and co-adapting to each other,” says Lane. “That seems to be genuinely difficult.” This improbability has implications for the search for alien life. On other worlds with the right chemical conditions, Lane believes that life would be sure to emerge. But without a fateful merger, it would be forever microbial. Perhaps this is the answer to the Fermi paradox—the puzzling contradiction between the high apparent odds that intelligent life would exist elsewhere among the billions of planets in the Milky Way, and our inability to find any signs of such intelligence. As Lane wrote in 2010, “The unavoidable conclusion is that the universe should be full of bacteria, but more complex life will be rare.” And if intelligent aliens did exist, they would probably have something like mitochondria, too. THE ORIGIN of eukaryotes is, by no means, a settled matter of fact. Ideas have waxed and waned in influence, and although many lines of evidence currently point to a sudden origin, there is still plenty of dissent. Some scientists support radical notions like the idea that prokaryotes are versions of eukaryotes that evolved to greater simplicity, rather than their ancestors. Others remain stalwart devotees of Woese’s tree. Writing in 2007, Anthony Poole and David Penny accused the sudden-origin camp of pushing “mechanisms founded in unfettered imagination.” They pointed out that archaea and bacteria do not engulf one another—that’s a hallmark of eukaryotes. It is easy to see how a primitive eukaryote might have gained mitochondria by engulfing a bacterium, but very hard to picture how a relatively simple archaeon did so. This powerful retort has lost some of its sting thanks to a white insect called the citrus mealybug. Its cells contain a bacterium called Tremblaya, and Tremblaya contains another bacterium called Moranella. Here is a prokaryote that somehow has another prokaryote living inside it, despite its apparent inability to engulf anything. Still, the details of how the initial archaeon-bacterium merger happened are still a mystery. How did one get inside the other? What sealed their partnership—was it hydrogen, as Martin and Müller suggested, or something else? How did they manage to stay conjoined? “I think we have the roadmap right, but we don’t have all the white lines and the signposts in place,” says Martin. “We have the big picture but not all the details.” Perhaps we will never know for sure. The origin of eukaryotes happened so far back in time that it’s a wonder we have even an inkling of what happened. Dissent is inevitable; uncertainty, guaranteed.
“You can’t convince everyone about anything in early evolution, because they hold to their own beliefs,” says Martin. “But I’m not worried about trying to convince anyone. I’ve solved these problems to my own satisfaction and it all looks pretty consistent. I’m happy.” ~ Ed Yong is an award-winning science writer. His work has appeared in Wired, Nature, the BBC, New Scientist, the Guardian, the Times, Aeon, Discover, Scientific American, The Scientist, the BMJ, Slate, and more. This article originally appeared in our “Mergers & Acquisitions” issue in February 2014.
The notion of junk DNA has always seemed like an oxymoron to me. Just because we don't know what it does (yet), does not mean it is junk. Apparently my view is shared by people who actually know a hell of lot more about genetics and DNA than do I. Still, there are even more cellular biologists who are not convinced.
The excellent article from Cosmos Magazine, by Dyani Lewis, looks at the current state of the field and the arguments from each side of the debate.
Scientists still argue whether the genome's 'dark matter' has any purpose. Dyani Lewis reports.
English geneticist Ewan Birney accepted a bet by his Australian colleague John Mattick that ‘junk DNA’ was our genome’s operating system. Mattick is still to collect. FAIRFAX
A little over a year ago it looked like Australian geneticist John Mattick had won a bet against his English colleague, Ewan Birney, over the way the human genome works. Like many others, Birney maintained that our genome was mostly comprised of “junk”, excess DNA that padded it out. Mattick, director of Sydney’s Garvan Institute, had long believed otherwise. In his view, so-called junk DNA would prove to be a code, our genome’s equivalent of a high-level operating system. In 2007 the two made a bet that at least 20% of the “junk” would be found to have a function. The stakes were a case of good Australian red. It was a well-timed wager. A worldwide project known as ENCODE was gearing up to examine the output of every one of the three billion letters of DNA that comprise the human genome. The results were announced in September 2012 with great fanfare. At a worldwide media conference, Birney declared that 80% of our DNA code was “functional”. Sometime, somewhere, one cell or another in the body was reading almost every bit of the genome. So can we call it quits on the debate over junk DNA? Far from it. As critics were quick to point out, simply reading out the DNA code is not proof that the code is functional. It might just be the cells’ equivalent of web surfing: a lot of useless sites get perused before anything useful is found. Mattick’s case of wine suddenly wasn’t looking quite such a sure thing.
John Rasko of the University of Sydney hates the term ‘junk DNA’. He believes it holds the key to how complex an organism is. CREDIT: JOHN RASKO
How to settle this argument? One way to decide whether junk DNA is useful would be to get rid of it and see what happens. Not an experiment you can do on people. But last year, Victor Albert at the State University of New York in Buffalo reported that nature might have done the experiment for us. Like us, the genomes of plants, insects and other animals also consist of vast amounts of DNA, much of which we can’t decipher. Albert claimed he had found a carnivorous plant, the bladderwort, which has a virtually junk-free genome and does just fine. Could the debate soon be settled? The term “junk DNA” was originally coined in 1972 by Japanese American evolutionary biologist Susumo Ohno. It’s easy to forget how little was known about genomes just four decades ago. In 1972 scientists could only speculate about what a whole genome might look like – how a four-letter DNA code of As, Ts, Gs and Cs might be strung together to write an instruction manual. But even without reading it, scientists knew that ours was big. The way Ohno saw it in the early 1970s, with a genome the size of ours, only a small percentage could possibly be made up of genes or we would suffer dangerous mutations that would quickly accrue over the generations. For decades, scientists focused on genes and ignored the junk. As many early geneticists found, if you mutate a gene, important developmental processes could be disrupted. At the time, a gene was thought of as a recipe for a protein. Proteins are the construction-site workers charged with turning the information in a one-dimensional DNA code into a living organism. They do it all, forming the bricks and mortar of our cells, the enzymes that drive our metabolism and the components of cell communications systems. But junk DNA could not be deciphered into any protein and the term became shorthand for any stretch of DNA that was not a protein-coding gene. Almost immediately the term seemed doomed. It was imprecise, and ignored growing evidence that some DNA sequences had other essential biological functions. For instance, researchers in the 1960s had already found that small tracts of DNA, known as “promoters”, lie directly ahead of protein-coding genes and act as helipads – landing sites for enzymes that read genes. These enzymes “transcribe” stretches of the DNA code into an almost identical stringy molecule called RNA. During the 1980s and 1990s, scientists managed to decipher even more novel functions for junk DNA. Other types of helipads, called “enhancers”, were identified, often located thousands of letters away from the gene they controlled. Yet other stretches of DNA carried instructions not for protein recipes, but for RNA recipes alone. Like a photocopy of a page from a recipe book, RNA was thought to be produced only for the purpose of instructing protein synthesis (see figure above). But it turns out that each transcribed RNA molecule could have a function. Some of these functional RNA molecules, dubbed “ribozymes”, work like enzymes to catalyse cellular reactions. Others, known as “microRNAs”, interfere with the RNA copies of other genes, effectively switching them off by preventing proteins from being made from the RNA recipe. Although these discoveries were momentous, they did not blow away the concept of junk. When the Human Genome Project finally unveiled its completed 3.2 billion letters of genetic code in 2003, the mystery of our un-deciphered genome hit prime time. The idea that only 1.5% of our DNA coded for genes seemed to fire the public imagination Our total number of genes was also humiliating. Ours was not the first genome to be unveiled: a microbe, a roundworm and a fruit fly all preceded us and revealed gene numbers ranging from 4,000 to 20,000. Surely our vastly more complex species would have at least an order of magnitude more. Not so. It turns out we have 20,000 protein-coding genes, the same number as the roundworm, a one millimetre long transparent creature boasting just 1,000 cells. “It was a great shock to everyone,” says University of Sydney haematologist John Rasko. Perhaps what set us apart from simpler organisms lay not in the genes, but in the 98.5% of our DNA still waiting to be decoded – the view firmly held by Mattick. He believes that the complexity of an organism does not relate to the number of genes, but to what’s in the junk DNA. Indeed there is a modest correlation between an organism’s complexity and the amount of junk DNA it carries: the bacterium E. coli contains little more than 10% non-protein coding DNA; roundworms 75%; for humans it’s 98.5%. Rasko hates the term “junk DNA”. “It still riles a lot of people in the field that the term ‘junk’ even took up traction,” he says. It’s not surprising that he is unimpressed with the phrase. Rasko’s “current obsession” is introns, the sort of DNA sequences Ohno would have dismissed as junk. Introns, as their name hints, are found interspersed within protein-coding genes and range in size from 10 to thousands of letters long. When a protein is made, the gene is first transcribed into an RNA copy with introns intact. But before the RNA molecule is finally translated into protein, the introns are edited out. Should that editing fail, the RNA molecule bearing an intact intron is sent to what Rasko calls “the molecular trash can” (see figure below). Rasko and his team have found that during the development of white blood cells, many RNA molecules actually hang on to their introns; a perplexing observation since these transcripts are made only to be trashed. “Why would a cell go to all of that trouble?” asks Rasko. The answer, he says, is “complexity”. Just as in the performance of a symphony orchestra, each instrument must play or be silent at precisely the right time, so too in the development of cells. Particular proteins need to be turned on and off at the stroke of a baton. By making transcripts that are destined for the shredder, Rasko believes that the genome has come up with “an elegant system” for orchestrating protein levels during the development of white blood cells. What’s more, entire suites of proteins can be orchestrated using the same molecular baton. Rasko identified 86 genes involved in white blood cell development that were all diminished in concert. And it turns out shredding the RNA instructions, rather than making unnecessary proteins, is much easier on the cell’s energy budget. “The energy costs on a cell by controlling the editing of introns are tens-fold less than it would be if you had to use a protein degradation mechanism,” he says. Introns are just one example of DNA sequences once viewed as superfluous, but now thought to be critical to the development of a complex organism such as a human. Disrupt intron editing and, as Rasko found, you disrupt the entire symphony. White blood cells unable to wield the baton failed to develop into the cells of the immune system. Rasko’s work illustrates how a once-overlooked component of the genome can turn out to be vital. The question is, how many other parts of the genome, once dubbed junk, are essential? That’s where ENCODE comes in. A small army of researchers joined forces in the wake of the Human Genome Project’s completion in 2003 to systematically sift through the vast tracts of mystery DNA. The purpose was to find which bits have a biological function. The massive international undertaking aimed to create the Encyclopaedia of DNA Elements (ENCODE’s full name) and brought together 442 scientists from around the globe. In September 2012, in an event that typifies the coordination required of such an immense project, their initial results were unveiled in a clutch of 30 scientific papers simultaneously published in three different scientific journals. The bottom line, as Birney – ENCODE’s lead analysis coordinator – announced to the media, is that 80% of the genome has a “biochemical function”. To arrive at this estimate, 147 types of cells were subjected to 24 different experiments to search for meaning in the oceans of DNA. What was surprising was the number of potentially useful sequences dotted throughout the genome. Instead of an immense ocean of junk DNA punctuated with occasional islands of protein-coding genes, the genome began to look like a thick soup, packed with active ingredients. Promoters and enhancers were known to be important residents of the mysterious non-coding DNA. But ENCODE found more than four million of them, many more than had previously been recognised. Combined with the 1.5% of protein-coding DNA, that takes the proportion of our genome with known function up to around 10%. ENCODE then measured other hints of function by looking at where proteins dock on to the long strands of DNA, finding three million of these sites. But the vast majority of “function” was inferred from the fact that in some cell somewhere in the body, at some time, DNA was being read, that is, transcribed into RNA.
Plant geneticist Jeffrey Bennetzen believes most DNA is useless. CREDIT: JEFFREY BENNETZEN
The ENCODE fanfare was answered with a storm of criticism. A “meaningless measure of functional significance”, tweeted Michael Eisen from the US Howard Hughes Medical Institute. The definition of “function” was “so loose as to be all but meaningless”, opined T. Ryan Gregory from the University of Guelph in Canada. The conclusions were “absurd” and full of “logical and methodological transgressions”, wrote Dan Graur from the University of Houston. Jeffrey Bennetzen, a plant geneticist from the University of Georgia, summarised the feeling: “I don’t think there’s anybody who believes that because something is transcribed, that means it has a function.” Mattick, who was involved in the pilot phase of ENCODE, disagrees. “I personally think it’s intellectually lazy to say it’s noisy transcription.” If it were noisy transcription, he says, then ENCODE would have seen random patterns of transcription. Instead it found precisely orchestrated patterns, tuned to particular cell types. Mattick believes that while gene number does not relate to complexity, those orchestrations of RNA transcribed from “junk” DNA do. As analysis of ENCODE continues, he predicts that the percentage of the human genome with proven function will edge towards 100%. For Magdalena Skipper, the editor at Nature who shepherded the publication of ENCODE’s Nature papers, arguments over the numbers are missing the point. “The value of ENCODE goes so much beyond this discussion of what is the percentage of the genome that is functional and in what way we define function.” No doubt. But we still want to know what most of our DNA is really doing. The answer might come from an unexpected place. The floating bladderwort is an unassuming carnivorous pondweed that captures its prey using tiny suction traps that lie beneath the water. But it wasn’t the bladderwort’s appearance or eating habits that intrigued evolutionary biologist Victor Albert. “It was known to have a tiny genome,” he says. “The question was, what’s missing?”
Albert and his colleagues found that the bladderwort genome contains a meagre 82 million letters. That’s 1/40 the size of our own, and an even punier 1/240 that of its plant relative, the Norway Spruce. But size was only half the story. “There’s essentially no junk DNA,” Albert says. The tiny genome contains around 28,500 protein-coding genes, but only 3% is what he would consider junk. “It’s an interesting counterpoint to the human genome situation.” Some have suggested that the bladderwort may have rid itself of excess DNA to save on phosphorous, an element that is part of the DNA molecule. Bladderworts live in an environment that is poor in phosphorous, and eat meat to bolster their intake of the element. (Albert himself doesn’t buy this explanation as to why they ditched their junk, since other phosphate-hungry carnivorous plants don’t have tiny genomes). So if the bladderwort can do all sorts of complex things without its excess genomic baggage, does it follow that junk DNA is irrelevant? Not necessarily. By “junk”, Albert was restricting his definition to a particular class of junk DNA known as “transposons” – repeating tracts that are relics of ancient viruses. And indeed the bladderwort seems to have dispensed with them. But as Mattick points out, even the minimalist bladderwort genome contains plenty of other non-protein coding sequences in the form of introns and tracts between genes that were traditionally termed junk – by his calculation some 65% of its genome. So, says Mattick, rather than spelling the death knell to junk, the bladderwort actually bolsters the view that no genome can truly go without. For Mattick, the bladderwort’s claim is just a replay of the claims made for the fugu, the highly poisonous Japanese puffer fish. For geneticists, it’s best known for having the tiniest genome of any back-boned animal, one-eighth the size of ours. When its genome was first read in 2002 it was similarly billed as a complex creature that had managed to do away with its “junk” DNA. But as Mattick points out, in fact 89% of fugu’s DNA does not code for proteins. So bladderworts and fugu still have a very high proportion of non-coding DNA, comparable to that of other complex organisms.
The carnivorous pondweed, bladderwort. The bladder trap, below, is an unusual structure that gives the plant its name. But a more surprising feature is the plant’s shrunken genome. CREDIT: GETTY IMAGES CREDIT: ENRIQUE IBARRA-LACLETTE, CLAUDIA ANAHÍ PÉREZ-TORRES AND PAULINA LOZANO-SOTOMAYOR
As for the transposons, the bits of old virus that seem to multiply in genomes, Mattick concedes that they could be padding the genomes of some plants. “But you don’t see nearly so much in animals,” he says, possibly because they are under greater evolutionary pressure than plants to streamline their genomes, keeping sequences that are useful, and jettisoning the rest. While no one argues that all non-protein coding DNA lacks function, the question now is how much is, in fact, junk? As Dan Graur cautions, when it comes to thinking about genomes, it’s a mistake to think in terms of a “Goldilocks genome” where every bit of DNA is perfectly fit for its function. “Evolution never breeds perfection,” he says. But even if a stretch of DNA is not perfectly functional, having some junk DNA to tinker with could be a big plus. As Mattick points out, bacteria with little “junk” have stayed stuck in the single-celled world whereas those with junk-laden genomes have formed the kingdoms of plants, animals and fungi. Perhaps genomes hang on to junk to allow the flexibility to evolve new and complex traits. But that loose association between junk DNA and complexity still doesn’t wash with many biologists. Until the function of the various sequences is demonstrated, biologists such as Albert, Bennetzen and Graur say that we are a long way from relegating the term “junk DNA” to the history books. Scientists such as Mattick and Rasko continue to pore over the “functional” DNA identified by ENCODE. But how much of the genome will eventually pass muster for the tougher critics is still open to wager. As geneticist Daniel MacArthur at Harvard University’s Broad Institute has declared, “I’d still take on Mattick’s wager any day, so long as I got to specify clearly what was meant by ‘functional’.”
Dyani Lewis is a freelance science journalist based in Melbourne, Australia.
This article came out in Pacific Standard back in September of last year, and I likely posted it then. But it turned up in tabs again recently and it still feels like an excellent and important article.
Your DNA is not a blueprint. Day by day, week by week, your genes are in a conversation with your surroundings. Your neighbors, your family, your feelings of loneliness: They don’t just get under your skin, they get into the control rooms of your cells. Inside the new social science of genetics.
•
A few years ago, Gene Robinson, of Urbana, Illinois, asked some associates in southern Mexico to help him kidnap some 1,000 newborns. For their victims they chose bees. Half were European honeybees, Apis mellifera ligustica, the sweet-tempered kind most beekeepers raise. The other half were ligustica’s genetically close cousins, Apis mellifera scutellata, the African strain better known as killer bees. Though the two subspecies are nearly indistinguishable, the latter defend territory far more aggressively. Kick a European honeybee hive and perhaps a hundred bees will attack you. Kick a killer bee hive and you may suffer a thousand stings or more. Two thousand will kill you. Working carefully, Robinson’s conspirators—researchers at Mexico’s National Center for Research in Animal Physiology, in the high resort town of Ixtapan de la Sal—jiggled loose the lids from two African hives and two European hives, pulled free a few honeycomb racks, plucked off about 250 of the youngest bees from each hive, and painted marks on the bees’ tiny backs. Then they switched each set of newborns into the hive of the other subspecies. Robinson, back in his office at the University of Illinois at Urbana-Champaign’s Department of Entomology, did not fret about the bees’ safety. He knew that if you move bees to a new colony in their first day, the colony accepts them as its own. Nevertheless, Robinson did expect the bees would be changed by their adoptive homes: He expected the killer bees to take on the European bees’ moderate ways and the European bees to assume the killer bees’ more violent temperament. Robinson had discovered this in prior experiments. But he hadn’t yet figured out how it happened. He suspected the answer lay in the bees’ genes. He didn’t expect the bees’ actual DNA to change: Random mutations aside, genes generally don’t change during an organism’s lifetime. Rather, he suspected the bees’ genes would behave differently in their new homes—wildly differently. This notion was both reasonable and radical. Scientists have known for decades that genes can vary their level of activity, as if controlled by dimmer switches. Most cells in your body contain every one of your 22,000 or so genes. But in any given cell at any given time, only a tiny percentage of those genes is active, sending out chemical messages that affect the activity of the cell. This variable gene activity, called gene expression, is how your body does most of its work. Sometimes these turns of the dimmer switch correspond to basic biological events, as when you develop tissues in the womb, enter puberty, or stop growing. At other times gene activity cranks up or spins down in response to changes in your environment. Thus certain genes switch on to fight infection or heal your wounds—or, running amok, give you cancer or burn your brain with fever. Changes in gene expression can make you thin, fat, or strikingly different from your supposedly identical twin. When it comes down to it, really, genes don’t make you who you are. Gene expression does. And gene expression varies depending on the life you live. Every biologist accepts this. That was the safe, reasonable part of Robinson’s notion. Where he went out on a limb was in questioning the conventional wisdom that environment usually causes fairly limited changes in gene expression. It might sharply alter the activity of some genes, as happens in cancer or digestion. But in all but a few special cases, the thinking went, environment generally brightens or dims the activity of only a few genes at a time. Robinson, however, suspected that environment could spin the dials on “big sectors of genes, right across the genome”—and that an individual’s social environment might exert a particularly powerful effect. Who you hung out with and how they behaved, in short, could dramatically affect which of your genes spoke up and which stayed quiet—and thus change who you were. Robinson was already seeing this in his bees. The winter before, he had asked a new post-doc, Cédric Alaux, to look at the gene-expression patterns of honeybees that had been repeatedly exposed to a pheromone that signals alarm. (Any honeybee that detects a threat emits this pheromone. It happens to smell like bananas. Thus “it’s not a good idea,” says Alaux, “to eat a banana next to a bee hive.”) To a bee, the pheromone makes a social statement: Friends, you are in danger. Robinson had long known that bees react to this cry by undergoing behavioral and neural changes: Their brains fire up and they literally fly into action. He also knew that repeated alarms make African bees more and more hostile. When Alaux looked at the gene-expression profiles of the bees exposed again and again to alarm pheromone, he and Robinson saw why: With repeated alarms, hundreds of genes—genes that previous studies had associated with aggression—grew progressively busier. The rise in gene expression neatly matched the rise in the aggressiveness of the bees’ response to threats. Robinson had not expected that. “The pheromone just lit up the gene expression, and it kept leaving it higher.” The reason soon became apparent: Some of the genes affected were transcription factors—genes that regulate other genes. This created a cascading gene-expression response, with scores of genes responding. This finding inspired Robinson’s kidnapping-and-cross-fostering study. Would moving baby bees to wildly different social environments reshape the curves of their gene-expression responses? Down in Ixtapan, Robinson’s collaborators suited up every five to 10 days, opened the hives, found about a dozen foster bees in each one, and sucked them up with a special vacuum. The vacuum shot them into a chamber chilled with liquid nitrogen. The intense cold instantly froze the bees’ every cell, preserving the state of their gene activity at that moment. At the end of six weeks, when the researchers had collected about 250 bees representing every stage of bee life, the team packed up the frozen bees and shipped them to Illinois. There, Robinson’s staff removed the bees’ sesame-seed-size brains, ground them up, and ran them through a DNA microarray machine. This identified which genes were busy in a bee’s brain at the moment it met the bee-vac. When Robinson sorted his data by group—European bees raised in African hives, for instance, or African bees raised normally among their African kin—he could see how each group’s genes reacted to their lives. Robinson organized the data for each group onto a grid of red and green color-coded squares: Each square represented a different gene, and its color represented the group’s average rate of gene expression. Red squares represented genes that were especially active in most of the bees in that group; the brighter the red, the more bees in which that gene had been busy. Green squares represented genes that were silent or underactive in most of the group. The printout of each group’s results looked like a sort of cubist Christmas card. When he got the cards, says Robinson, “the results were stunning.” For the bees that had been kidnapped, life in a new home had indeed altered the activity of “whole sectors” of genes. When their gene expression data was viewed on the cards alongside the data for groups of bees raised among their own kin, a mere glance showed the dramatic change. Hundreds of genes had flipped colors. The move between hives didn’t just make the bees act differently. It made their genes work differently, and on a broad scale. What’s more, the cards for the adopted bees of both species came to ever more resemble, as they moved through life, the cards of the bees they moved in with. With every passing day their genes acted more like those of their new hive mates (and less like those of their genetic siblings back home). Many of the genes that switched on or off are known to affect behavior; several are associated with aggression. The bees also acted differently. Their dispositions changed to match that of their hive mates. It seemed the genome, without changing its code, could transform an animal into something very like a different subspecies. These bees didn’t just act like different bees. They’d pretty much become different bees. To Robinson, this spoke of a genome far more fluid—far more socially fluid—than previously conceived.
Gene Robinson, an entomologist at the University of Illinois, found that when European honeybees are raised among more aggressive African killer bees, they not only start to become as belligerent as their new hive mates—they come to genetically resemble them. (PHOTO: COURTESY OF GENE ROBINSON)
ROBINSON SOON REALIZED HE was not alone in seeing this. At conferences and in the literature, he kept bumping into other researchers who saw gene networks responding fast and wide to social life. David Clayton, a neurobiologist also on the University of Illinois campus, found that if a male zebra finch heard another male zebra finch singing nearby, a particular gene in the bird’s forebrain would “re up—and it would do so differently depending on whether the other finch was strange and threatening, or familiar and safe. Others found this same gene, dubbed ZENK ramping up in other species. In each case, the change in ZENK’s activity corresponded to some change in behavior: a bird might relax in response to a song, or become vigilant and tense. Duke researchers, for instance, found that when female zebra finches listened to male zebra finches’ songs, the females’ ZENK gene triggered massive gene-expression changes in their forebrains—a socially sensitive brain area in birds as well as humans. The changes differed depending on whether the song was a mating call or a territorial claim. And perhaps most remarkably, all of these changes happened incredibly fast—within a half hour, sometimes within just five minutes. ZENK, it appeared, was a so-called “immediate early gene,” a type of regulatory gene that can cause whole networks of other genes to change activity. These sorts of regulatory gene-expression response had already been identified in physiological systems such as digestion and immunity. Now they also seemed to drive quick responses to social conditions. One of the most startling early demonstrations of such a response occurred in 2005 in the lab of Stanford biologist Russell Fernald. For years, Fernald had studied the African cichlid Astatotilapia burtoni, a freshwater fish about two inches long and dull pewter in color. By 2005 he had shown that among burtoni, the top male in any small population lives like some fishy pharaoh, getting far more food, territory, and sex than even the No. 2 male. This No. 1 male cichlid also sports a bigger and brighter body. And there is always only one No. 1. I wonder, Fernald thought, what would happen if we just removed him? So one day Fernald turned out the lights over one of his cichlid tanks, scooped out big flashy No. 1, and then, 12 hours later, flipped the lights back on. When the No. 2 cichlid saw that he was now No. 1, he responded quickly. He underwent massive surges in gene expression that immediately blinged up his pewter coloring with lurid red and blue streaks and, in a matter of hours, caused him to grow some 20 percent. It was as if Jason Schwartzman, coming to work one day to learn the big office stud had quit, morphed into Arnold Schwarzenegger by close of business. These studies, says Greg Wray, an evolutionary biologist at Duke who has focused on gene expression for over a decade, caused quite a stir. “You suddenly realize birds are hearing a song and having massive, widespread changes in gene expression in just 15 minutes? Something big is going on.” This big something, this startlingly quick gene-expression response to the social world, is a phenomenon we are just beginning to understand. The recent explosion of interest in “epigenetics”—a term literally meaning “around the gene,” and referring to anything that changes a gene’s effect without changing the actual DNA sequence—has tended to focus on the long game of gene-environment interactions: how famine among expectant mothers in the Netherlands during World War II, for instance, affected gene expression and behavior in their children; or how mother rats, by licking and grooming their pups more or less assiduously, can alter the wrappings around their offspring’s DNA in ways that influence how anxious the pups will be for the rest of their lives. The idea that experience can echo in our genes across generations is certainly a powerful one. But to focus only on these narrow, long-reaching effects is to miss much of the action where epigenetic influence and gene activity is concerned. This fresh work by Robinson, Fernald, Clayton, and others—encompassing studies of multiple organisms, from bees and birds to monkeys and humans—suggests something more exciting: that our social lives can change our gene expression with a rapidity, breadth, and depth previously overlooked. Why would we have evolved this way? The most probable answer is that an organism that responds quickly to fast-changing social environments will more likely survive them. That organism won’t have to wait around, as it were, for better genes to evolve on the species level. Immunologists discovered something similar 25 years ago: Adapting to new pathogens the old-fashioned way—waiting for natural selection to favor genes that create resistance to specific pathogens—would happen too slowly to counter the rapidly changing pathogen environment. Instead, the immune system uses networks of genes that can respond quickly and flexibly to new threats. We appear to respond in the same way to our social environment. Faced with an unpredictable, complex, ever-changing population to whom we must respond successfully, our genes behave accordingly—as if a fast, fluid response is a matter of life or death. ABOUT THE TIME ROBINSON was seeing fast gene expression changes in bees, in the early 2000s, he and many of his colleagues were taking notice of an up-and-coming UCLA researcher named Steve Cole. Cole, a Californian then in his early 40s, had trained in psychology at the University of California-Santa Barbara and Stanford; then in social psychology, epidemiology, virology, cancer, and genetics at UCLA. Even as an undergrad, Cole had “this astute, fine-grained approach,” says Susan Andersen, a professor of psychology now at NYU who was one of his teachers at UC Santa Barbara in the late 1980s. “He thinks about things in very precise detail.” In his post-doctoral work at UCLA, Cole focused on the genetics of immunology and cancer because those fields had pioneered hard-nosed gene-expression research. After that, he became one of the earliest researchers to bring the study of whole-genome gene-expression to social psychology. The gene’s ongoing, real-time response to incoming information, he realized, is where life works many of its changes on us. The idea is both reductive and expansive. We are but cells. At each cell’s center, a tight tangle of DNA writes and hands out the cell’s marching orders. Between that center and the world stand only a series of membranes. “Porous membranes,” notes Cole. “We think of our bodies as stable biological structures that live in the world but are fundamentally separate from it. That we are unitary organisms in the world but passing through it. But what we’re learning from the molecular processes that actually keep our bodies running is that we’re far more fluid than we realize, and the world passes through us.” Cole told me this over dinner. We had met on the UCLA campus and walked south a few blocks, through bright April sun, to an almost empty sushi restaurant. Now, waving his chopsticks over a platter of urchin, squid, and amberjack, he said, “Every day, as our cells die off, we have to replace one to two percent of our molecular being. We’re constantly building and re-engineering new cells. And that regeneration is driven by the contingent nature of gene expression. “This is what a cell is about. A cell,” he said, clasping some amberjack, “is a machine for turning experience into biology.” When Cole started his social psychology research in the early 1990s, the microarray technology that spots changes in gene expression was still in its expensive infancy, and saw use primarily in immunology and cancer. So he began by using the tools of epidemiology—essentially the study of how people live their lives. Some of his early papers looked at how social experience affected men with HIV. In a 1996 study of 80 gay men, all of whom had been HIV-positive but healthy nine years earlier, Cole and his colleagues found that closeted men succumbed to the virus much more readily. He then found that HIV-positive men who were lonely also got sicker sooner, regardless of whether they were closeted. Then he showed that closeted men without HIV got cancer and various infectious diseases at higher rates than openly gay men did. At about the same time, psychologists at Carnegie Mellon finished a well-controlled study showing that people with richer social ties got fewer common colds. Something about feeling stressed or alone was gumming up the immune system—sometimes fatally. “You’re besieged by a virus that’s going to kill you,” says Cole, “but the fact that you’re socially stressed and isolated seems to shut down your viral defenses. What’s going on there?” He was determined to find out. But the research methods on hand at the time could take him only so far: “Epidemiology won’t exactly lie to you. But it’s hard to get it to tell you the whole story.” For a while he tried to figure things out at the bench, with pipettes and slides and assays. “I’d take norepinephrine [a key stress hormone] and squirt it on some infected T-cells and watch the virus grow faster. The norepinephrine was knocking down the antiviral response. That’s great. Virologists love that. But it’s not satisfying as a complete answer, because it doesn’t fully explain what’s happening in the real world. “You can make almost anything happen in a test tube. I needed something else. I had set up all this theory. I needed a place to test it.” His next step was to turn to rhesus monkeys, a lab species that allows controlled study. In 2007, he joined John Capitanio, a primatologist at the University of California-Davis, in looking at how social stress affected rhesus monkeys with SIV, or simian immunodeficiency virus, the monkey version of HIV. Capitanio had found that monkeys with SIV fell ill and died faster if they were stressed out by constantly being moved into new groups among strangers—a simian parallel to Cole’s 1996 study on lonely gay men. Capitanio had run a rough immune analysis that showed the stressed monkeys mounted weak antiviral responses. Cole offered to look deeper. First he tore apart the lymph nodes—“ground central for infection”—and found that in the socially stressed monkeys, the virus bloomed around the sympathetic nerve trunks, which carry stress signals into the lymph node. “This was a hint,” says Cole: The virus was running amok precisely where the immune response should have been strongest. The stress signals in the nerve trunks, it seemed, were getting either muted en route or ignored on arrival. As Cole looked closer, he found it was the latter: The monkeys’ bodies were generating the appropriate stress signals, but the immune system didn’t seem to be responding to them properly. Why not? He couldn’t find out with the tools he had. He was still looking at cells. He needed to look inside them. Finally Cole got his chance. At UCLA, where he had been made a professor in 2001, he had been working hard to master gene-expression analysis across an entire genome. Microarray machines—the kind Gene Robinson was using on his bees—were getting cheaper. Cole got access to one and put it to work. Thus commenced what we might call the lonely people studies. First, in collaboration with University of Chicago social psychologist John Cacioppo, Cole mined a questionnaire about social connections that Cacioppo had given to 153 healthy Chicagoans in their 50s and 60s. Cacioppo and Cole identified the eight most socially secure people and the six loneliest and drew blood samples from them. (The socially insecure half-dozen were lonely indeed; they reported having felt distant from others for the previous four years.) Then Cole extracted genetic material from the blood’s leukocytes (a key immune-system player) and looked at what their DNA was up to. He found a broad, weird, strongly patterned gene-expression response that would become mighty familiar over the next few years. Of roughly 22,000 genes in the human genome, the lonely and not-lonely groups showed sharply different gene-expression responses in 209. That meant that about one percent of the genome—a considerable portion—was responding differently depending on whether a person felt alone or connected. Printouts of the subjects’ gene-expression patterns looked much like Robinson’s red-and-green readouts of the changes in his cross-fostered bees: Whole sectors of genes looked markedly different in the lonely and the socially secure. And many of these genes played roles in inflammatory immune responses. Now Cole was getting somewhere. Normally, a healthy immune system works by deploying what amounts to a leashed attack dog. It detects a pathogen, then sends inflammatory and other responses to destroy the invader while also activating an anti-inflammatory response—the leash—to keep the inflammation in check. The lonely Chicagoans’ immune systems, however, suggested an attack dog off leash—even though they weren’t sick. Some 78 genes that normally work together to drive inflammation were busier than usual, as if these healthy people were fighting infection. Meanwhile, 131 genes that usually cooperate to control inflammation were underactive. The underactive genes also included key antiviral genes. This opened a whole new avenue of insight. If social stress reliably created this gene-expression profile, it might explain a lot about why, for instance, the lonely HIV carriers in Cole’s earlier studies fell so much faster to the disease. But this was a study of just 14 people. Cole needed more. Over the next several years, he got them. He found similarly unbalanced gene-expression or immune-response profiles in groups including poor children, depressed people with cancer, and people caring for spouses dying of cancer. He topped his efforts off with a study in which social stress levels in young women predicted changes in their gene activity six months later. Cole and his collaborators on that study, psychologists Gregory Miller and Nicolas Rohleder of the University of British Columbia, interviewed 103 healthy Vancouver-area women aged 15 to 19 about their social lives, drew blood, and ran gene-expression profiles, and after half a year drew blood and ran profiles again. Some of the women reported at the time of the initial interview that they were having trouble with their love lives, their families, or their friends. Over the next six months, these socially troubled subjects took on the sort of imbalanced gene-expression profile Cole found in his other isolation studies: busy attack dogs and broken leashes. Except here, in a prospective study, he saw the attack dog breaking free of its restraints: Social stress changed these young women’s gene-expression patterns before his eyes.
Gene-expression microarray printouts (this one comes from a study of autistic versus non-autistic people) depict snapshots of activity across a genome. Red squares represent genes that are more active, green squares represent genes that are less active. (PHOTO: PUBLIC DOMAIN)
IN EARLY 2009, COLE sat down to make sense of all this in a review paper that he would publish later that year in Current Directions in Psychological Science. Two years later we sat in his spare, rather small office at UCLA and discussed what he’d found. Cole, trimly built but close to six feet tall, speaks in a reedy voice that is slightly higher than his frame might lead you to expect. Sometimes, when he’s grabbing for a new thought or trying to emphasize a point, it jumps a register. He is often asked to give talks about his work, and it’s easy to see why: Relaxed but animated, he speaks in such an organized manner that you can almost see the paragraphs form in the air between you. He spends much of his time on the road. Thus the half-unpacked office, he said, gesturing around him. His lab, down the hall, “is essentially one really good lab manager”—Jesusa M. Arevalo, whom he frequently lists on his papers—“and a bunch of robots,” the machines that run the assays. “We typically think of stress as being a risk factor for disease,” said Cole. “And it is, somewhat. But if you actually measure stress, using our best available instruments, it can’t hold a candle to social isolation. Social isolation is the best-established, most robust social or psychological risk factor for disease out there. Nothing can compete.” This helps explain, for instance, why many people who work in high-stress but rewarding jobs don’t seem to suffer ill effects, while others, particularly those isolated and in poverty, wind up accruing lists of stress-related diagnoses—obesity, Type 2 diabetes, hypertension, atherosclerosis, heart failure, stroke. Despite these well-known effects, Cole said he was amazed when he started finding that social connectivity wrought such powerful effects on gene expression. “Or not that we found it,” he corrected, “but that we’re seeing it with such consistency. Science is noisy. I would’ve bet my eyeteeth that we’d get a lot of noisy results that are inconsistent from one realm to another. And at the level of individual genes that’s kind of true—there is some noise there.” But the kinds of genes that get dialed up or down in response to social experience, he said, and the gene networks and gene-expression cascades that they set off, “are surprisingly consistent—from monkeys to people, from five-year-old kids to adults, from Vancouver teenagers to 60-year-olds living in Chicago.” COLE’S WORK CARRIES ALL kinds of implications—some weighty and practical, some heady and philosophical. It may, for instance, help explain the health problems that so often haunt the poor. Poverty savages the body. Hundreds of studies over the past few decades have tied low income to higher rates of asthma, flu, heart attacks, cancer, and everything in between. Poverty itself starts to look like a disease. Yet an empty wallet can’t make you sick. And we all know people who escape poverty’s dangers. So what is it about a life of poverty that makes us ill? Cole asked essentially this question in a 2008 study he conducted with Miller and Edith Chen, another social psychologist then at the University of British Columbia. The paper appeared in an odd forum: Thorax, a journal about medical problems in the chest. The researchers gathered and ran gene-expression profiles on 31 kids, ranging from nine to 18 years old, who had asthma; 16 were poor, 15 well-off. As Cole expected, the group of well-off kids showed a healthy immune response, with elevated activity among genes that control pulmonary inflammation. The poorer kids showed busier inflammatory genes, sluggishness in the gene networks that control inflammation, and—in their health histories—more asthma attacks and other health problems. Poverty seemed to be mucking up their immune systems. Cole, Chen, and Miller, however, suspected something else was at work—something that often came with poverty but was not the same thing. So along with drawing the kids’ blood and gathering their socioeconomic information, they showed them films of ambiguous or awkward social situations, then asked them how threatening they found them. The poorer kids perceived more threat; the well-off perceived less. This difference in what psychologists call “cognitive framing” surprised no one. Many prior studies had shown that poverty and poor neighborhoods, understandably, tend to make people more sensitive to threats in ambiguous social situations. Chen in particular had spent years studying this sort of effect. But in this study, Chen, Cole, and Miller wanted to see if they could tease apart the effect of cognitive framing from the effects of income disparity. It turned out they could, because some of the kids in each income group broke type. A few of the poor kids saw very little menace in the ambiguous situations, and a few well-off kids saw a lot. When the researchers separated those perceptions from the socioeconomic scores and laid them over the gene-expression scores, they found that it was really the kids’ framing, not their income levels, that accounted for most of the difference in gene expression. To put it another way: When the researchers controlled for variations in threat perception, poverty’s influence almost vanished. The main thing driving screwy immune responses appeared to be not poverty, but whether the child saw the social world as scary. But where did that come from? Did the kids see the world as frightening because they had been taught to, or because they felt alone in facing it? The study design couldn’t answer that. But Cole believes isolation plays a key role. This notion gets startling support from a 2004 study of 57 school-age children who were so badly abused that state social workers had removed them from their homes. The study, often just called “the Kaufman study,” after its author, Yale psychiatrist Joan Kaufman, challenges a number of assumptions about what shapes responses to trauma or stress. The Kaufman study at first looks like a classic investigation into the so-called depression risk gene—the serotonin transporter gene, or SERT—which comes in both long and short forms. Any single gene’s impact on mood or behavior is limited, of course, and these single-gene, or “candidate gene,” studies must be viewed with that in mind. Yet many studies have found that SERT’s short form seems to render many people (and rhesus monkeys) more sensitive to environment; according to those studies, people who carry the short SERT are more likely to become depressed or anxious if faced with stress or trauma. Kaufman looked first to see whether the kids’ mental health tracked their SERT variants. It did: The kids with the short variant suffered twice as many mental-health problems as those with the long variant. The double whammy of abuse plus short SERT seemed to be too much. Then Kaufman laid both the kids’ depression scores and their SERT variants across the kids’ levels of “social support.” In this case, Kaufman narrowly defined social support as contact at least monthly with a trusted adult figure outside the home. Extraordinarily, for the kids who had it, this single, modest, closely defined social connection erased about 80 percent of the combined risk of the short SERT variant and the abuse. It came close to inoculating kids against both an established genetic vulnerability and horrid abuse. Or, to phrase it as Cole might, the lack of a reliable connection harmed the kids almost as much as abuse did. Their isolation wielded enough power to raise the question of what’s really most toxic in such situations. Most of the psychiatric literature essentially views bad experiences—extreme stress, abuse, violence—as toxins, and “risk genes” as quasi-immunological weaknesses that let the toxins poison us. And abuse is clearly toxic. Yet if social connection can almost completely protect us against the well-known effects of severe abuse, isn’t the isolation almost as toxic as the beatings and neglect? The Kaufman study also challenges much conventional Western thinking about the state of the individual. To use the language of the study, we sometimes conceive of “social support” as a sort of add-on, something extra that might somehow fortify us. Yet this view assumes that humanity’s default state is solitude. It’s not. Our default state is connection. We are social creatures, and have been for eons. As Cole’s colleague John Cacioppo puts it in his book Loneliness, Hobbes had it wrong when he wrote that human life without civilization was “solitary, poor, nasty, brutish, and short.” It may be poor, nasty, brutish, and short. But seldom has it been solitary. TOWARD THE END OF the dinner I shared with Cole, after the waiter took away the empty platters and we sat talking over green tea, I asked him if there was anything I should have asked but had not. He’d been talking most of three hours. Some people run dry. Cole does not. He spoke about how we are permeable fluid beings instead of stable unitary isolates; about recursive reconstruction of the self; about an engagement with the world that constantly creates a new you, only you don’t know it, because you’re not the person you would have been otherwise—you’re a one-person experiment that has lost its control. He wanted to add one more thing: He didn’t see any of this as deterministic. We were obviously moving away from what he could prove at this point, perhaps from what is testable. We were in fact skirting the rabbit hole that is the free-will debate. Yet he wanted to make it clear he does not see us as slaves to either environment or genes. “You can’t change your genes. But if we’re even half right about all this, you can change the way your genes behave—which is almost the same thing. By adjusting your environment you can adjust your gene activity. That’s what we’re doing as we move through life. We’re constantly trying to hunt down that sweet spot between too much challenge and too little. “That’s a really important part of this: To an extent that immunologists and psychologists rarely appreciate, we are architects of our own experience. Your subjective experience carries more power than your objective situation. If you feel like you’re alone even when you’re in a room filled with the people closest to you, you’re going to have problems. If you feel like you’re well supported even though there’s nobody else in sight; if you carry relationships in your head; if you come at the world with a sense that people care about you, that you’re valuable, that you’re okay; then your body is going to act as if you’re okay—even if you’re wrong about all that.” Cole was channeling John Milton: “The mind is its own place, and in itself can make a heaven of hell, a hell of heaven.” Of course I did not realize that at the moment. My reaction was more prosaic. “So environment and experience aren’t the same,” I offered. “Exactly. Two people may share the same environment but not the same experience. The experience is what you make of the environment. It appears you and I are both enjoying ourselves here, for instance, and I think we are. But if one of us didn’t like being one-on-one at a table for three hours, that person could get quite stressed out. We might have much different experiences. And you can shape all this by how you frame things. You can shape both your environment and yourself by how you act. It’s really an opportunity.” Cole often puts it differently at the end of his talks about this line of work. “Your experiences today will influence the molecular composition of your body for the next two to three months,” he tells his audience, “or, perhaps, for the rest of your life. Plan your day accordingly.”